Highly Cited Papers
Our group was founded in 1988 by Klaus Schulten. His publications with the group (1988 - 2016) have been cited over 56,000 times (ISI, August 2015) and all of his publications (1972 - 2016) have been cited over 92,000 times (Google citation profile) as of December 2016. The hundred most highly cited publications are listed below with descriptions of the associated scientific discoveries or technology developments.
 VMD - Visual
Molecular Dynamics. William Humphrey, Andrew Dalke, and Klaus Schulten.
Journal of Molecular Graphics, 14:33-38,
1996.
VMD - Visual
Molecular Dynamics. William Humphrey, Andrew Dalke, and Klaus Schulten.
Journal of Molecular Graphics, 14:33-38,
1996. (Citations: 19,441) VMD is a molecular graphics program designed for the display and analysis of molecular assemblies, in particular biopolymers such as proteins and nucleic acids. VMD can simultaneously display any number of structures using a wide variety of rendering styles and coloring methods. Molecules are displayed as one or more `representations', where each representation embodies a particular rendering method and coloring scheme for a selected subset of atoms. The atoms displayed in each representation are chosen using an extensive atom selection syntax, which includes boolean operators and regular expressions. VMD provides a complete graphical user interface for program control, as well as a text interface using the Tcl embeddable parser to allow for complex scripts with variable substitution, control loops, and function calls. Full session logging is supported, which produces a VMD command script for later playback. High-resolution raster images of displayed molecules may be produced by generating input scripts for use by a number of photorealistic image processing applications. VMD has also been expressly designed with the ability to animate molecular dynamics (MD) simulation trajectories, imported either from files or from a direct connection to a running MD simulation. VMD is the visualization component of MDScope, a set of tools for interactive problem solving in structural biology, which also includes the parallel MD program NAMD, and the MDCOMM software used to connect the visualization and simulation programs. VMD is written in C++, using an object-oriented design; the program, including source code and extensive documentation, is freely available via anonymous ftp and through the World Wide Web. |
 Scalable molecular
dynamics with NAMD. James C. Phillips, Rosemary Braun, Wei Wang, James
Gumbart, Emad Tajkhorshid, Elizabeth Villa, Christophe Chipot, Robert D.
Skeel, Laxmikant Kale, and Klaus Schulten. Journal of Computational Chemistry,
26:1781-1802, 2005.
Scalable molecular
dynamics with NAMD. James C. Phillips, Rosemary Braun, Wei Wang, James
Gumbart, Emad Tajkhorshid, Elizabeth Villa, Christophe Chipot, Robert D.
Skeel, Laxmikant Kale, and Klaus Schulten. Journal of Computational Chemistry,
26:1781-1802, 2005. (Citations: 8,118) NAMD is a parallel molecular dynamics code designed for high-performance simulation of large biomolecular systems. NAMD scales to hundreds of processors on high-end parallel platforms, as well as tens of processors on low-cost commodity clusters, and also runs on individual desktop and laptop computers. NAMD works with AMBER and CHARMM potential functions, parameters, and file formats. This article, directed to novices as well as experts, first introduces concepts and methods used in the NAMD program, describing the classical molecular dynamics force field, equations of motion, and integration methods along with the efficient electrostatics evaluation algorithms employed and temperature and pressure controls used. Features for steering the simulation across barriers and for calculating both alchemical and conformational free energy differences are presented. The motivations for and a roadmap to the internal design of NAMD, implemented in C++ and based on Charm++ parallel objects, are outlined. The factors affecting the serial and parallel performance of a simulation are discussed. Finally, typical NAMD use is illustrated with representative applications to a small, a medium, and a large biomolecular system, highlighting particular features of NAMD, for example, the Tcl scripting language. The article also provides a list of the key features of NAMD and discusses the benefits of combining NAMD with the molecular graphics/sequence analysis software VMD and the grid computing/collaboratory software BioCoRE. NAMD is distributed free of charge with source code at www.ks.uiuc.edu. |
 NAMD2: Greater scalability for parallel molecular dynamics. Laxmikant Kalé, Robert Skeel, Milind Bhandarkar, Robert Brunner,
Attila Gursoy, Neal Krawetz, James Phillips, Aritomo
Shinozaki, Krishnan Varadarajan, and Klaus Schulten. Journal of Computational
Physics, 151:283-312, 1999.
NAMD2: Greater scalability for parallel molecular dynamics. Laxmikant Kalé, Robert Skeel, Milind Bhandarkar, Robert Brunner,
Attila Gursoy, Neal Krawetz, James Phillips, Aritomo
Shinozaki, Krishnan Varadarajan, and Klaus Schulten. Journal of Computational
Physics, 151:283-312, 1999. (Citations: 2,021) Molecular dynamics programs simulate the behavior of biomolecular systems, leading to insights and understanding of their functions. However, the computational complexity of such simulations is enormous. Parallel machines provide the potential to meet this computational challenge. To harness this potential, it is necessary to develop a scalable program. It is also necessary that the program be easily modified by application-domain programmers. The NAMD2 program presented in this paper seeks to provide these desirable features. It uses spatial decomposition combined with force decomposition to enhance scalability. It uses intelligent periodic load balancing, so as to maximally utilize the available compute power. It is modularly organized, and implemented using a parallel C++ dialect, so as to enhance its modifiability. It uses a combination of numerical techniques and algorithms to ensure that energy drifts are minimized, ensuring accuracy in long running calculations. NAMD2 uses a portable run-time framework that also supports interoperability among multiple parallel paradigms. As a result, different components of applications can be written in the most appropriate parallel paradigms. NAMD2 runs on most parallel machines including workstation clusters. This paper also describes the performance obtained on some benchmark applications. |
 "Neural gas" for vector quantization and its application to time-series prediction. Thomas M. Martinetz, Stanislav G.
Berkovich, and Klaus Schulten. IEEE Transactions on Neural Networks,
4:558-569, 1993.
"Neural gas" for vector quantization and its application to time-series prediction. Thomas M. Martinetz, Stanislav G.
Berkovich, and Klaus Schulten. IEEE Transactions on Neural Networks,
4:558-569, 1993. (Citations: 1,428) As a data compression technique, vector quantization requires the minimalization of a cost function - the distortion error - which, in general, has many local minima. In this paper, a neural network algorithm based on a "soft-max" adaptation rule is presented that exhibits good performance in reaching the optimum, or at least coming close. The soft-max rule employed is an extension of the standard K-means clustering procedure and takes into account a "neighborhood ranking" of the reference (weight) vectors. It is shown that the dynamics of the reference (weight) vectors during the input-driven adaptation procedure 1) is determined by the gradient of an energy function whose shape can be modulated through a neighborhood determining parameter, and 2) resembles the dynamics of Brownian particles moving in a potential determined by the data point density. The network is employed to represent the attractor of the Mackey-Glass equation and to predict the Mackey-Glass time series, with additional local linear mappings for generating output values. The results obtained for the time-series prediction compare very favorably with the results achieved by back-propagation and radial basis function networks. |
 Molecular biomimetics: nanotechnology through biology. Mehmet Sarikaya, Candan Tamerler, Alex K. -Y. Jen, Klaus Schulten,
and François Baneyx. Nature Materials, 2:577-585, 2003.
Molecular biomimetics: nanotechnology through biology. Mehmet Sarikaya, Candan Tamerler, Alex K. -Y. Jen, Klaus Schulten,
and François Baneyx. Nature Materials, 2:577-585, 2003. (Citations: 1,386) Proteins, through their unique and specific interactions with other macromolecules and inorganics, control structures and functions of all biological hard and soft tissues in organisms. Molecular biomimetics is an emerging field in which hybrid technologies are developed by using the tools of molecular biology and nanotechnology. Taking lessons from biology, polypeptides can now be genetically engineered to specifically bind to selected inorganic compounds for applications in nano- and biotechnology. This review discusses combinatorial biological protocols, that is, bacterial cell surface and phage-display technologies, in the selection of short sequences that have affinity to (noble) metals, semiconducting oxides and other technological compounds. These genetically engineered proteins for inorganics (GEPIs) can be used in the assembly of functional nanostructures. Based on the three fundamental principles of molecular recognition, self-assembly and DNA manipulation, we highlight successful uses of GEPI in nanotechnology. |
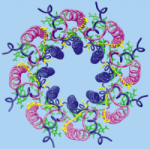 The crystal structure of the light harvesting complex II (B800-850) from Rhodospirillum molischianum. Juergen Koepke, Xiche
Hu, Cornelia Muenke, Klaus Schulten, and Hartmut Michel.
Structure, 4:581-597, 1996.
The crystal structure of the light harvesting complex II (B800-850) from Rhodospirillum molischianum. Juergen Koepke, Xiche
Hu, Cornelia Muenke, Klaus Schulten, and Hartmut Michel.
Structure, 4:581-597, 1996. (Citations: 1,023) In photosynthesis light is absorbed by light-harvesting antenna complexes (LHs) and its energy is transferred to the photosynthetic reaction center. In purple photosynthetic bacteria and higher plants the LHs are integral membrane protein/pigment complexes. LH-II from the purple bacterium Rhodospirillum molischianum is an octamer of heterodimers, the later consisting of two polypeptides, the |
 Textbook: Neural
Computation and Self-Organizing Maps: An Introduction. Helge Ritter,
Thomas Martinetz, and Klaus Schulten. Addison-Wesley, New York, revised
English edition, 1992.
Textbook: Neural
Computation and Self-Organizing Maps: An Introduction. Helge Ritter,
Thomas Martinetz, and Klaus Schulten. Addison-Wesley, New York, revised
English edition, 1992. (Citations: 1,004) This book is a comprehensive introduction to neural networks and neural information processing. It describes the most important models of neural networks and how they contribute to our understanding of information and organization processes in the brain. One of the few generally recognized organizational principles of the nervous system, the development of cortical feature maps (brain maps), is described in detail, and the reader is introduced to the biological background and the mathematical properties of self-organizing maps as important functional building blocks of the brain. Examples show how neural networks can solve important information processing tasks, including the development of sensory maps, the traveling salesman problem, and visuomotor control of robots. |
 Topology
representing networks. Thomas Martinetz and Klaus Schulten. Neural
Networks, 7:507-522, 1994.
Topology
representing networks. Thomas Martinetz and Klaus Schulten. Neural
Networks, 7:507-522, 1994. (Citations: 922) A Hebbian adaptation rule with winner-take-all like competition is introduced. It is shown that this competitive Hebbian rule forms so-called Delaunay triangulations, which play an important role in computational geometry for efficiently solving proximity problems. Given a set of neural units i, i = 1, ..., N, the synaptic weights of which can be interpreted as pointers |
 A "neural gas" network learns topologies.
Thomas Martinetz and Klaus Schulten.
In Teuvo Kohonen, Kai Mäkisara, Olli Simula, and Jari Kangas, editors, Artificial Neural Networks, pp. 397-402. Elsevier, Amsterdam,
A "neural gas" network learns topologies.
Thomas Martinetz and Klaus Schulten.
In Teuvo Kohonen, Kai Mäkisara, Olli Simula, and Jari Kangas, editors, Artificial Neural Networks, pp. 397-402. Elsevier, Amsterdam, (Citations: 897) A neural network algorithm for vector quantization of topologically arbitrarily structured manifolds of input signals is presented and applied to a data manifold M which consists of subsets of different dimensionalities. In addition to the quantization of M each neural unit i, i + 1, ..., N of the network A develops connections, described by |
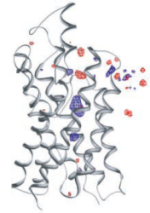 Control of the selectivity of the aquaporin water channel family by global orientational tuning. Emad Tajkhorshid, Peter
Nollert, Morten Ø. Jensen, Larry J. W. Miercke, Joseph O'Connell,
Robert M. Stroud, and Klaus Schulten. Science, 296:525-530, 2002.
Control of the selectivity of the aquaporin water channel family by global orientational tuning. Emad Tajkhorshid, Peter
Nollert, Morten Ø. Jensen, Larry J. W. Miercke, Joseph O'Connell,
Robert M. Stroud, and Klaus Schulten. Science, 296:525-530, 2002. (Citations: 730) Aquaporins are transmembrane channels found in cell membranes of all life forms. We examine their apparently paradoxical property, facilitation of efficient permeation of water while excluding protons, which is of critical importance to preserving the electrochemical potential across the cell membrane. We have determined the structure of the Escherichia coli aquaglyceroporin GlpF with bound water, in native (2.7 angstroms) and in W48F/F200T mutant (2.1 angstroms) forms, and carried out 12-nanosecond molecular dynamics simulations that define the spatial and temporal probability distribution and orientation of a single file of seven to nine water molecules inside the channel. Two conserved asparagines force a central water molecule to serve strictly as a hydrogen bond donor to its neighboring water molecules. Assisted by the electrostatic potential generated by two half-membrane spanning loops, this dictates opposite orientations of water molecules in the two halves of the channel, and thus prevents the formation of a "proton wire," while permitting rapid water diffusion. Both simulations and observations revealed a more regular distribution of channel water and an increased water permeability for the W48F/F200T mutant. |
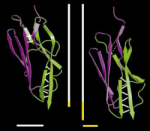 Mechanical unfolding intermediates in titin modules. Piotr E. Marszalek, Hui Lu, Hongbin Li, Mariano Carrion-Vazquez, Andres
F. Oberhauser, Klaus Schulten, and Julio M. Fernandez. Nature,
402:100-103, 1999.
Mechanical unfolding intermediates in titin modules. Piotr E. Marszalek, Hui Lu, Hongbin Li, Mariano Carrion-Vazquez, Andres
F. Oberhauser, Klaus Schulten, and Julio M. Fernandez. Nature,
402:100-103, 1999. (Citations: 721) The modular protein titin, which is responsible for the passive elasticity of muscle, is subjected to stretching forces. Previous work on the experimental elongation of single titin molecules has suggested that force causes consecutive unfolding of each domain in an all-or-none fashion |
 A model for photoreceptor-based magnetoreception in birds. Thorsten Ritz, Salih Adem, and Klaus Schulten. Biophysical
Journal, 78:707-718, 2000.
A model for photoreceptor-based magnetoreception in birds. Thorsten Ritz, Salih Adem, and Klaus Schulten. Biophysical
Journal, 78:707-718, 2000. (Citations: 678) A large variety of animals has the ability to sense the geomagnetic field and utilize it as a source of directional (compass) information. It is not known by which biophysical mechanism this magnetoreception is achieved. We investigate the possibility that magnetoreception involves radical pair processes which are governed by anisotropic hyperfine coupling between (unpaired) electron and nuclear spins. We will show theoretically that fields of geomagnetic field strength and weaker can produce significantly different reaction yields for different alignments of the radical pairs with the magnetic field. As a model for a magnetic sensory organ we propose a system of radical pairs being (1) orientationally ordered in a molecular substrate and (2) exhibiting changes in the reaction yields that affect the visual transduction pathway. We evaluate three-dimensional visual modulation patterns that can arise from the influence of the geomagnetic field on radical pair systems. The variations of these patterns with orientation and field strength can furnish the magnetic compass ability of birds with the same characteristics as observed in behavioral experiments. We propose that the recently discovered photoreceptor cryptochrome is part of the magnetoreception system and suggest further studies to prove or disprove this hypothesis. |
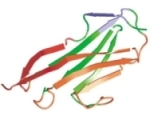 Steered molecular dynamics and mechanical functions of proteins. Barry Isralewitz, Mu Gao, and Klaus Schulten. Current
Opinion in Structural Biology, 11:224-230, 2001.
Steered molecular dynamics and mechanical functions of proteins. Barry Isralewitz, Mu Gao, and Klaus Schulten. Current
Opinion in Structural Biology, 11:224-230, 2001. (Citations: 646) Atomic force microscopy of single molecules, steered molecular dynamics, and the theory of stochastic processes have established a new field that investigates mechanical functions of proteins such as ligand - receptor binding/unbinding and elasticity of muscle proteins during stretching. The combination of these methods yields information on the energy landscape that controls mechanical function and on the force bearing components of proteins, as well as on the underlying physical mechanisms. |
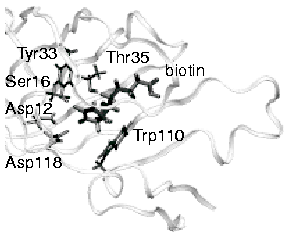 Molecular
Dynamics Study of Unbinding of the Avidin-Biotin Complex Sergei
Izrailev, Sergey Stepaniants, Manel Balsera, Yoshi Oono, and Klaus
Schulten. Biophysical Journal, 72:1568-1581, 1997.
Molecular
Dynamics Study of Unbinding of the Avidin-Biotin Complex Sergei
Izrailev, Sergey Stepaniants, Manel Balsera, Yoshi Oono, and Klaus
Schulten. Biophysical Journal, 72:1568-1581, 1997. (Citations: 631) We report molecular dynamics simulations which induce, over periods of 40-500 ps, the unbinding of biotin from avidin by means of external harmonic forces with force constraints close to those of AFM cantilevers. The applied forces are sufficiently large to reduce the overall binding energy enough to yield unbinding within the measurement time. Our study complements earlier work on biotin-streptavidin that employed a much larger harmonic force constant. The simulations reveal a variety of unbinding pathways, the role of key residues contributing to adhesion as well as the spatial range over which avidin binds biotin. In contrast to the previous studies, the calculated rupture forces exceed by far those observed. We demonstrate, in the framework of models expressed in terms of one-dimensional Langevin equations with a schematic binding potential, the associated Smoluchowski equations, and the theory of first passage times, that picosecond to nanosecond simulation of ligand unbinding requires such strong forces that the resulting protein-ligand motion proceeds far from the thermally activated regime of millisecond AFM experiments, and that simulated unbinding cannot be readily extrapolated to the experimentally observed rupture. |
 Accelerating molecular modeling applications with graphics processors.
John E. Stone, James C. Phillips, Peter L. Freddolino, David J. Hardy, Leonardo G. Trabuco, and Klaus Schulten.
Journal of Computational Chemistry, 28:2618-2640, 2007.
Accelerating molecular modeling applications with graphics processors.
John E. Stone, James C. Phillips, Peter L. Freddolino, David J. Hardy, Leonardo G. Trabuco, and Klaus Schulten.
Journal of Computational Chemistry, 28:2618-2640, 2007. (Citations: 623) Molecular mechanics simulations offer a computational approach to studying the behavior of biomolecules at atomic detail, but such simulations are limited in size and timescale by available computing resources. State-of-the-art graphics processing units (GPUs) can perform over 500 billion arithmetic operations per second, a tremendous computational resource which can now be utilized for general purpose computing as a result of recent advances in GPU hardware and software architecture. In this paper, an overview of recent advances in programmable GPUs is presented, with an emphasis on their application to molecular mechanics simulations and the programming techniques required to obtain optimal performance in these cases. We demonstrate the use of GPUs for the calculation of long-range electrostatics and nonbonded forces for molecular dynamics simulations, where GPU-based calculations are typically 10-100 times faster than heavily optimized CPU-based implementations. The application of GPU acceleration to biomolecular simulation is also demonstrated through the use of GPU-accelerated Coulomb-based ion placement and calculation of time-averaged potentials from molecular dynamics trajectories. A novel approximation to Coulomb potential calculation, the multilevel summation method, is introduced and compared to direct Coulomb summation. In light of the performance obtained for this set of calculations, future applications of graphics processors to molecular dynamics simulations are discussed. |
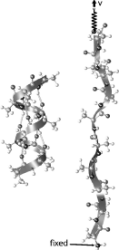
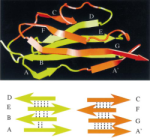 Unfolding of titin immunoglobulin domains by steered molecular dynamics simulation. Hui Lu, Barry Isralewitz, André Krammer,
Viola Vogel, and Klaus Schulten. Biophysical Journal,
75:662-671, 1998.
Unfolding of titin immunoglobulin domains by steered molecular dynamics simulation. Hui Lu, Barry Isralewitz, André Krammer,
Viola Vogel, and Klaus Schulten. Biophysical Journal,
75:662-671, 1998. (Citations: 612) Titin, a 1 |
 First passage time approach to diffusion controlled reactions. Attila Szabo, Klaus Schulten, and Zan Schulten. Journal of
Chemical Physics, 72:4350-4357, 1980.
First passage time approach to diffusion controlled reactions. Attila Szabo, Klaus Schulten, and Zan Schulten. Journal of
Chemical Physics, 72:4350-4357, 1980.(Citations: 611) Association reactions involving diffusion in one, two, and three-dimensional finite domains governed by Smoluchowski-type equations (e.g., interchain reaction of macromolecules, ligand binding to receptors, repressor-operator association of DNA strand) are shown to be often well described by first-order kinetics and characterized by an average reaction (passage) time |
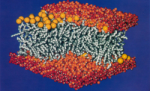 Molecular dynamics simulation of a bilayer of 200 lipids in the gel and in the liquid crystal-phases. Helmut Heller,
Michael Schaefer, and Klaus Schulten. Journal of Physical Chemistry,
97:8343-8360, 1993.
Molecular dynamics simulation of a bilayer of 200 lipids in the gel and in the liquid crystal-phases. Helmut Heller,
Michael Schaefer, and Klaus Schulten. Journal of Physical Chemistry,
97:8343-8360, 1993. (Citations: 527) We have constructed and simulated a membrane-water system which consists of 200 molecules of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine forming a rectangular patch of a bilayer and of 5483 water molecules covering the head groups on each side of the bilayer. The total number of atoms is approximately 27 000. The lateral dimensions of the bilayer are 85 A |
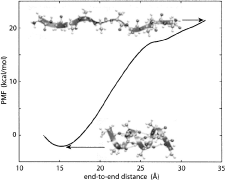 Calculating
potentials of mean force from steered molecular dynamics simulations.
Sanghyun Park and Klaus Schulten. Journal of Chemical Physics, 120:5946-5961,
2004.
Calculating
potentials of mean force from steered molecular dynamics simulations.
Sanghyun Park and Klaus Schulten. Journal of Chemical Physics, 120:5946-5961,
2004. (Citations: 498) Steered molecular dynamics (SMD) permits efficient investigations of molecular processes by focusing on selected degrees of freedom. We explain how one can, in the framework of SMD, employ Jarzynski's equality (also known as the nonequilibrium work relation) to calculate potentials of mean force (PMF). We outline the theory that serves this purpose and connects nonequilibrium processes (such as SMD simulations) with equilibrium properties (such as the PMF). We review the derivation of Jarzynski's equality, generalize it to isobaric-isothermal processes, and discuss its implications in relation to the second law of thermodynamics and computer simulations. In the relevant regime of steering by means of stiff springs, we demonstrate that the work on the system is Gaussian-distributed regardless of the speed of the process simulated. In this case, the cumulant expansion of Jarzynski's equality can be safely terminated at second order. We illustrate the PMF calculation method for an exemplary simulation and demonstrate the Gaussian nature of the resulting work distribution. |
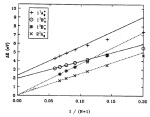
 Free energy
calculation from steered molecular dynamics simulations using Jarzynski's
equality.Sanghyun Park, Fatemeh Khalili-Araghi, Emad Tajkhorshid, and
Klaus Schulten. Journal of Chemical Physics, 119:3559-3566, 2003.
Free energy
calculation from steered molecular dynamics simulations using Jarzynski's
equality.Sanghyun Park, Fatemeh Khalili-Araghi, Emad Tajkhorshid, and
Klaus Schulten. Journal of Chemical Physics, 119:3559-3566, 2003. (Citations: 486) Jarzynski's equality is applied to free energy calculations from steered molecular dynamics simulations of biomolecules. The helix-coil transition of deca-alanine in vacuum is used as an example. With about ten trajectories sampled, the second order cumulant expansion, among the various averaging schemes examined, yields the most accurate estimates. We compare umbrella sampling and the present method, and find that their efficiencies are comparable. |
 Electronic
excitations in finite and infinite polyenes. Paul Tavan and Klaus
Schulten. Physical Review B, 36:4337-4358,
1987.
Electronic
excitations in finite and infinite polyenes. Paul Tavan and Klaus
Schulten. Physical Review B, 36:4337-4358,
1987. (Citations: 480) We study electronic excitations in long polyenes, i.e., in one-dimensional strongly correlated electron systems which are neither infinite nor small. The excitations are described within Hubbard and Pariser-Parr-Pople (PPP) models by means of a multiple-reference double-excitation expansion [P. Tavan and K. Schulten, J. Chem. Phys. 85, 6602 (1986)]. We find that quantized "transition" momenta can be assigned to electronic excitations in finite chains. These momenta link excitation energies of finite chains to dispersion relations of infinite chains, i.e., they bridge the gap between finite and infinite systems. A key result is the following: Excitation energies E in polyenes with N carbon atoms are described very accurately by the formula |
 Linear polyene
electronic structure and potential surfaces. Bruce S. Hudson, Bryan E.
Kohler, and Klaus Schulten. In Edward C. Lim, editor, Excited States, volume
6, pp. 1-95. Academic Press, New York, 1982.
Linear polyene
electronic structure and potential surfaces. Bruce S. Hudson, Bryan E.
Kohler, and Klaus Schulten. In Edward C. Lim, editor, Excited States, volume
6, pp. 1-95. Academic Press, New York, 1982. (Citations: 462) Polyenes are linear conjugated chains of carbon atoms joined by alternating double and single bonds and are deservedly the objects of a good deal of experimental and theoretical attention. There are many reasons for this, including the historical importance of these systems in the development of molecular quantum theory, the fundamental importance of cis-trans photoisomerization, which is the distinctive photochemistry of these molecules, and the fact that polyene chromophores play starring roles in biologically important photoprocesses, such as vision and energy production in the purple-membrane Halobacterium halobium. In both of these biological examples, the electronic structure of teh polyene and how it changes upon excitation is of key importance. Until relatively recently, this structure was thought to be rather simple, similar to that of other conjugated systems such as the polyacenes, and well described by approximate molecular orbital ideas. Recent experiments and theoretical discoveries have revealed that this is not the case: Polyene electronic structure is both more complicated and more interesting than was previously thought. |
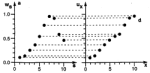 Self-organizing
maps: Ordering, convergence properties and energy functions. Edgar Erwin,
Klaus Obermayer, and Klaus Schulten. Biological Cybernetics, 67:47-55, 1992.
Self-organizing
maps: Ordering, convergence properties and energy functions. Edgar Erwin,
Klaus Obermayer, and Klaus Schulten. Biological Cybernetics, 67:47-55, 1992.
(Citations: 448) We investigate the convergence properties of the self-organizing feature map algorithm for a simple, but very instructive case: the formation of a topographic representation of the unit interval [0,1] by a linear chain of neurons. We extend the proofs of convergence of Kohonen and of Cottrell and Fort to hold in any case where the neighborhood function, which is used to scale the change in the weight values at each neuron, is a monotonically decreasing function of distance from the winner neuron. We prove that the learning dynamics cannot be described by a gradient descent on a single energy function, but may be described using a set of potential functions, one for each neuron, which are independently minimized following a stochastic gradient descent. We derive the correct potential functions for the one- and multi-dimensional case, and show that the energy functions given by Tolat (1990) are an approximation which is no longer valid in the case of highly disordered maps or steep neighborhood functions. |
 NAMD - A parallel, object-oriented molecular dynamics program.
Mark Nelson, William Humphrey, Attila Gursoy, Andrew Dalke, Laxmikant Kalé,Robert D. Skeel, and Klaus Schulten.
International Journal of Supercomputer Applications and High Performance
Computing, 10:251-268, 1996.
NAMD - A parallel, object-oriented molecular dynamics program.
Mark Nelson, William Humphrey, Attila Gursoy, Andrew Dalke, Laxmikant Kalé,Robert D. Skeel, and Klaus Schulten.
International Journal of Supercomputer Applications and High Performance
Computing, 10:251-268, 1996.(Citations: 442) NAMD is a molecular dynamics program designed for high performance simulations of large biomolecular systems on parallel computers. An object-oriented design implemented using C++ facilitates the incorporation of new algorithms into the program. NAMD uses spatial decomposition coupled with a multithreaded, message-driven design which is shown to scale efficiently to multiple processors. Also, NAMD incorporates the Distributed Parallel Multipole Tree Algorithm for computation of full electrostatic force evaluation in O(N) time. NAMD can be connected via a communication system to a molecular graphics program in order to provide an interactive modeling tool for viewing and modifying a running simulation. The application of NAMD to a protein-DNA -water complex of more 36,000 atoms illustrates the performance of NAMD. |
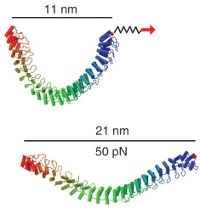 Single-molecule experiments in vitro and in silico. Marcos Sotomayor and Klaus Schulten. Science, 316:1144-1148, 2007 Single-molecule experiments in vitro and in silico. Marcos Sotomayor and Klaus Schulten. Science, 316:1144-1148, 2007 (Citations: 439) Single-molecule force experiments in vitro enable the characterization of the mechanical response of biological matter at the nanometer scale. However, they do not reveal the molecular mechanisms underlying mechanical function. These can only be readily studied through molecular dynamics simulations of atomic structural models: 'in silico' (by computer analysis) single-molecule experiments. Steered molecular dynamics simulations, in which external forces are used to explore the response and function of macromolecules, have become a powerful tool complementing and guiding in vitro single-molecule experiments. The insights provided by in silico experiments are illustrated here through a review of recent research in three areas of protein mechanics: elasticity of the muscle protein titin and the extracellular matrix protein fibronectin; linker-mediated elasticity of the cytoskeleton protein spectrin; and elasticity of ankyrin repeats, a protein module found ubiquitously in cells but with an as-yet unclear function. |
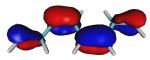 On the origin of a low-lying forbidden transition in polyenes and related molecules. Klaus Schulten and Martin Karplus.
Chemical Physics Letters, 14:305-309, 1972.
On the origin of a low-lying forbidden transition in polyenes and related molecules. Klaus Schulten and Martin Karplus.
Chemical Physics Letters, 14:305-309, 1972. (Citations: 422) It is demonstrated that the inclusion of double-excited configurations in semi-empirical and a priori calculations of polyenes leads to a significant alteration of the spectrum. In agreement with the recent experiment of Hudson and Kohler, a forbidden ( |
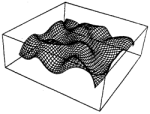 Convergence properties of Kohonen's topology conserving maps: Fluctuations, stability and dimension selection. Helge Ritter
and Klaus Schulten. Biological Cybernetics, 60:59-71, 1988.
Convergence properties of Kohonen's topology conserving maps: Fluctuations, stability and dimension selection. Helge Ritter
and Klaus Schulten. Biological Cybernetics, 60:59-71, 1988. (Citations: 406) We analyse a Markovian algorithm for the formation of topologically correct feature maps proposed earlier by Kohonen. The maps from a space of input signals onto an array of formal neurons are generated by a learning scheme driven by a random sequence of input samples. The learning is described by an equivalent Fokker-Planck equation. Convergence to an equilibrium map can be ensured by a criterion for the time dependence of the learning step size. We investigate the stability of the equilibrium map and calculate the fluctuations around it. We also study an instability responsible for a phenomenon termed by Kohonen "automatic selection of feature dimensions". |
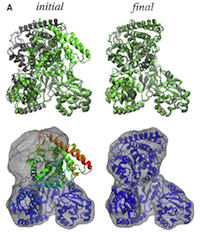 Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Leonardo G. Trabuco, Elizabeth Villa,
Kakoli Mitra, Joachim Frank, and Klaus Schulten. Structure, 16:673-683, 2008.
Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Leonardo G. Trabuco, Elizabeth Villa,
Kakoli Mitra, Joachim Frank, and Klaus Schulten. Structure, 16:673-683, 2008. (Citations: 398) A novel method to flexibly fit atomic structures into electron microscopy (EM) maps using molecular dynamics simulations is presented. The simulations incorporate the EM data as an external potential added to the molecular dynamics force field, allowing all internal features present in the EM map to be used in the fitting process, while the model remains fully flexible and stereochemically correct. The molecular dynamics flexible fitting (MDFF) method is validated for available crystal structures of protein and RNA in different conformations; measures to assess and monitor the fitting process are introduced. The MDFF method is then used to obtain high-resolution structures of the E. coli ribosome in different functional states imaged by cryo-EM. |
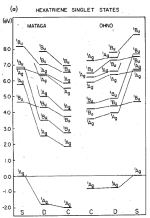 Correlation effects in the spectra of polyenes. Klaus Schulten, I. Ohmine, and Martin Karplus. Journal of Chemical Physics,
64:4422-4441, 1976.
Correlation effects in the spectra of polyenes. Klaus Schulten, I. Ohmine, and Martin Karplus. Journal of Chemical Physics,
64:4422-4441, 1976. (Citations: 394) A Hamiltonian of the Pariser-Parr-Pople form is employed to investigate the effect of correlation on the |
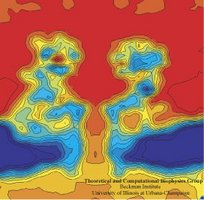 Imaging
alpha-hemolysin with molecular dynamics: Ionic conductance, osmotic
permeability and the electrostatic potential map. Aleksij Aksimentiev and Klaus Schulten. Biophysical Journal, 88:3745-3761, 2005 Imaging
alpha-hemolysin with molecular dynamics: Ionic conductance, osmotic
permeability and the electrostatic potential map. Aleksij Aksimentiev and Klaus Schulten. Biophysical Journal, 88:3745-3761, 2005 (Citations: 390) |
 Architecture and Mechanism of the light harvesting apparatus of purple bacteria. Xiche Hu, Ana Damjanovic, Thorsten Ritz, and
Klaus Schulten. Proceedings of the National Academy of
Sciences, USA, 95:5935-5941, 1998. (Citations: 390)
Architecture and Mechanism of the light harvesting apparatus of purple bacteria. Xiche Hu, Ana Damjanovic, Thorsten Ritz, and
Klaus Schulten. Proceedings of the National Academy of
Sciences, USA, 95:5935-5941, 1998. (Citations: 390)Photosynthetic organisms fuel their metabolism with light energy and have developed for this purpose an efficient apparatus for harvesting sunlight. The atomic structure of the apparatus, as it evolved in purple bacteria, has been constructed through a combination of X-ray crystallography, electron microscopy, and modeling. The detailed structure and overall architecture reveals a hierarchical aggregate of pigments that utilizes, as shown through femtosecond spectroscopy and quantum physics, elegant and efficient mechanisms for primary light absorption and transfer of electronic excitation towards the photosynthetic reaction center. |
 Generalized Verlet algorithm for efficient molecular dynamics simulations with long-range interactions.
Helmut Grubmüller, Helmut Heller, Andreas Windemuth, and Klaus Schulten.
Molecular Simulation, 6:121-142, 1991.
Generalized Verlet algorithm for efficient molecular dynamics simulations with long-range interactions.
Helmut Grubmüller, Helmut Heller, Andreas Windemuth, and Klaus Schulten.
Molecular Simulation, 6:121-142, 1991. (Citations: 386) For the purpose of molecular dynamics simulations of large biopolymers we have developed a new method to accelerate the calculation of long-range pair interactions (e.g. Coulomb interaction). The algorithm introduces distance classes to schedule updates of non-bonding interactions and to avoid unnecessary computations of interactions between particles which are far apart. To minimize the error caused by the updating schedule, the Verlet integration scheme has been modified. The results of the method are compared to those of other approximation schemes as well as to results obtained by numerical integration without approximation. For simulation of a protein with 12 637 atoms our approximation scheme yields a reduction of computer time by a factor of seven. The approximation suggested can be implemented on sequential as well as on parallel computers. We describe an implementation on a (Transputer-based) MIMD machine with a systolic ring architecture. |
 Molecular dynamics simulations of the complete satellite tobacco mosaic virus.
Peter L. Freddolino, Anton S. Arkhipov, Steven B. Larson, Alexander McPherson, and Klaus Schulten.
Structure, 14:437-449, 2006.
Molecular dynamics simulations of the complete satellite tobacco mosaic virus.
Peter L. Freddolino, Anton S. Arkhipov, Steven B. Larson, Alexander McPherson, and Klaus Schulten.
Structure, 14:437-449, 2006. (Citations: 371) This work presents an all-atom molecular dynamics simulation of a complete virus, the satellite tobacco mosaic virus. Simulations with up to 1 million atoms for over 50 ns demonstrate the stability of the whole virion and of the RNA core alone, while the capsid without RNA exhibits a pronounced instability. Physical properties of the simulated virus particle including electrostatic potential, radial distribution of viral components, and patterns of correlated motion are analyzed and the implications for the assembly and infection mechanism of the virus are discussed. |
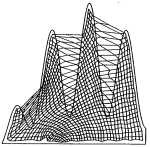 On the stationary state of Kohonen's self-organizing sensory mapping. Helge Ritter and Klaus Schulten. Biological
Cybernetics,
54:99-106, 1986.
On the stationary state of Kohonen's self-organizing sensory mapping. Helge Ritter and Klaus Schulten. Biological
Cybernetics,
54:99-106, 1986. (Citations: 366) The stationary state of the self-organizing sensory mappings of Kohonen is investigated. For this purpose the equation for the stationary state is derived for the case of one-dimensional and two-dimensional mappings. The equation can be solved for special cases, including the general one-dimensional case, to yield an explicit expression for the local magnification factor of the map. |
 Photosynthetic apparatus of purple bacteria. Xiche Hu, Thorsten Ritz, Ana Damjanovic, Felix Autenrieth, and Klaus Schulten.
Quarterly Reviews of Biophysics, 35:1-62, 2002.
Photosynthetic apparatus of purple bacteria. Xiche Hu, Thorsten Ritz, Ana Damjanovic, Felix Autenrieth, and Klaus Schulten.
Quarterly Reviews of Biophysics, 35:1-62, 2002. (Citations: 339) This article reviews work accomplished during the past decade on the structure and function of the photosynthetic unit of purple bacteria with a main focus on the light harvesting component. The photosynthetic unit exists as aggregates of proteins in the intracellular membranes of these bacteria; the units absorb sun light and utilize its energy for the synthesis of ATP. The light harvesting component involves ring-shape proteins that surround directly in the form of satellite rings the so-called reaction center. The structure of the proteins as established through a combination of experimental and computational methods is reviewed. The proteins provide a scaffold for a hierarchical aggregate of chlorophylls and carotenoids that funnel electronic excitation towards the reaction center. The physics of this process is reviewed in detail. Finally, the genomic level organization of the light harvesting system is summarized. |
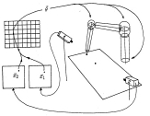 Topology-conserving maps for learning visuo-motor-coordination. Helge Ritter, Thomas Martinetz, and Klaus Schulten. Neural Networks, 2:159-168, 1989 Topology-conserving maps for learning visuo-motor-coordination. Helge Ritter, Thomas Martinetz, and Klaus Schulten. Neural Networks, 2:159-168, 1989 (Citations: 324) We investigate the application of an extension of Kohonen's self-organizing mapping algorithm to the learning of visuo-motor-coordination of a simulated robot arm. We show that both arm kinematics and arm dynamics can be learned, if a suitable representation for the map output is used. Due to the topology-conserving property of the map spatially neighboring neurons can learn cooperatively, which greatly improves the robustness and the convergence properties of the algorithm. |
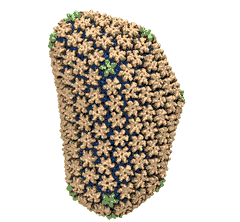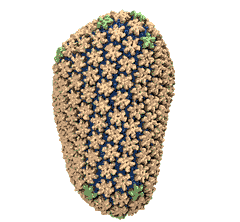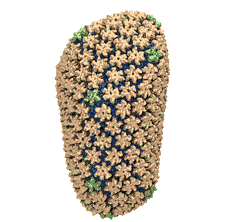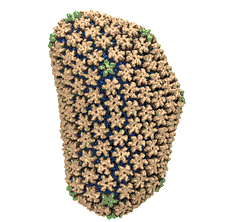 Mature HIV-1 capsid
structure by cryo-electron microscopy and all-atom molecular dynamics. Gongpu Zhao, Juan R. Perilla, Ernest L. Yufenyuy, Xin Meng, Bo Chen, Jiying Ning, Jinwoo Ahn, Angela M. Gronenborn, Klaus Schulten, Christopher Aiken, and Peijun Zhang.
Nature, 497:643-646,
2013.
Mature HIV-1 capsid
structure by cryo-electron microscopy and all-atom molecular dynamics. Gongpu Zhao, Juan R. Perilla, Ernest L. Yufenyuy, Xin Meng, Bo Chen, Jiying Ning, Jinwoo Ahn, Angela M. Gronenborn, Klaus Schulten, Christopher Aiken, and Peijun Zhang.
Nature, 497:643-646,
2013. (Citations: 320) Human immunodeficiency virus type 1 (HIV-1) is the major cause of AIDS, for which treatments need to be developed continuously as the virus becomes quickly resistant to new drugs. When the virus infects a human cell it releases into the cell its capsid, a closed, stable container protecting the viral genetic material. However, interaction with the cell triggers at some point an instability of the capsid, leading to a well timed release of the genetic material that merges then with the cell's genes and begins to control the cell. The dual role of the capsid, to be functionally both stable and unstable, makes it in principle an ideal target for antiviral drugs and, in fact, treatments of other viral infections successfully target the respective capsids. The size of the HIV-1 capsid (about 1,300 proteins), and its irregular shape had prevented so far the resolution of a full capsid atomic-level structure. However, in a tour de force effort, groups of experimental and computational scientists have now resolved the capsid's chemical structure (deposited to the protein data bank under the accession codes 3J3Q and 3J3Y). The discovery can guide now the design of novel drugs for enhanced antiviral therapy. |
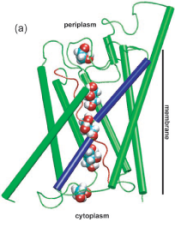 Energetics of glycerol conduction through aquaglyceroporin GlpF. Morten Ø. Jensen, Sanghyun Park,
Emad Tajkhorshid, and Klaus Schulten. Proceedings of the National Academy
of Sciences, USA, 99:6731-6736, 2002.
Energetics of glycerol conduction through aquaglyceroporin GlpF. Morten Ø. Jensen, Sanghyun Park,
Emad Tajkhorshid, and Klaus Schulten. Proceedings of the National Academy
of Sciences, USA, 99:6731-6736, 2002. (Citations: 320) Aquaglyceroporin GlpF selectively conducts water and linear polyalcohols, such as glycerol, across the inner membrane of Escherichia coli. We report steered molecular dynamics simulations of glycerol conduction through GlpF, in which external forces accelerate the transchannel conduction in a manner that preserves the intrinsic conduction mechanism. The simulations reveal channel-glycerol hydrogen bonding interactions and the stereoselectivity of the channel. Employing Jarzynski's identity between free energy and irreversible work, we reconstruct the potential of mean force along the conduction pathway through a time series analysis of molecular dynamics trajectories. This potential locates binding sites and barriers inside the channel; it also reveals a low energy periplasmic vestibule suited for efficient uptake of glycerol from the environment. |
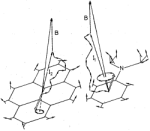 Semiclassical
description of electron spin motion in radicals including the effect of
electron hopping. Klaus Schulten and Peter G. Wolynes. Journal of Chemical
Physics, 68:3292-3297, 1978.
Semiclassical
description of electron spin motion in radicals including the effect of
electron hopping. Klaus Schulten and Peter G. Wolynes. Journal of Chemical
Physics, 68:3292-3297, 1978.(Citations: 320) The coherent electron spin motion in radicals induced by the hyperfine coupling to nuclear spins is described semiclassically. The nuclear spins are treated as constant classical vectors around which the electron spin precesses. The ensemble average over all nuclear spin configurations is taken yielding the electron spin correlation tensor |
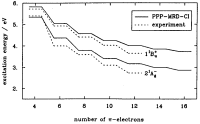 The low-lying
electronic excitations in long polyenes: A PPP-MRD-CI study. Paul Tavan
and Klaus Schulten. Journal of Chemical Physics, 85:6602-6609, 1986.
The low-lying
electronic excitations in long polyenes: A PPP-MRD-CI study. Paul Tavan
and Klaus Schulten. Journal of Chemical Physics, 85:6602-6609, 1986.
(Citations: 319) A correct description of the electronic excitations in polyenes demands that electron correlation is accounted for correctly. Very large expansions are necessary including many electron configurations with at least one, two, three, and four electrons promoted from the Hartree-Fock ground state. The enormous size of such expansions had prohibited accurate computations of the spectra for polyenes with more than ten |
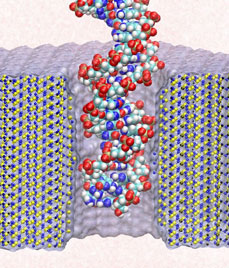 Sizing DNA
using a nanometer-diameter pore. J. B. Heng, C. Ho, T. Kim, R. Timp, A. Aksimentiev, Y. V. Grinkova, S. Sligar, K. Schulten, and G. Timp. Biophysical Journal, 87:2905-2911, 2004 Sizing DNA
using a nanometer-diameter pore. J. B. Heng, C. Ho, T. Kim, R. Timp, A. Aksimentiev, Y. V. Grinkova, S. Sligar, K. Schulten, and G. Timp. Biophysical Journal, 87:2905-2911, 2004 (Citations: 315) Each species from bacteria to human has a distinct genetic fingerprint. Therefore, a mechanism that detects a single molecule of DNA represents the ultimate analytical tool. As a first step in the development of such a tool, we have explored using a nanometer- diameter pore, sputtered in a nanometer-thick inorganic membrane with a tightly focused electron beam, as a transducer that detects single molecules of DNA and produces an electrical signature of the structure. When an electric field is applied across the membrane,. DNA molecule immersed in electrolyte is attracted to the pore, blocks the current through it, and eventually translocates across the membrane as verified by gel electrophoresis. The relationship between DNA translocation and blocking current signature has been established through molecular dynamics simulations. By measuring the duration and the magnitude of the blocking current transient, we can discriminate single-stranded from |
 Textbook: Neuronale Netze: Eine Einführung in die Neuroinformatik
selbstorganisierender Abbildungen.
Helge Ritter, Thomas Martinetz, and Klaus Schulten.
Addison-Wesley, Bonn, second enlarged edition, 1990.
Textbook: Neuronale Netze: Eine Einführung in die Neuroinformatik
selbstorganisierender Abbildungen.
Helge Ritter, Thomas Martinetz, and Klaus Schulten.
Addison-Wesley, Bonn, second enlarged edition, 1990. (Citations: 314) This book is a comprehensive introduction to neural networks and neural information processing. It describes the most important models of neural networks and how they contribute to our understanding of information and organization processes in the brain. One of the few generally recognized organizational principles of the nervous system, the development of cortical feature maps (brain maps), is described in detail, and the reader is introduced to the biological background and the mathematical properties of self-organizing maps as important functional building blocks of the brain. Examples show how neural networks can solve important information processing tasks, including the development of sensory maps, the traveling salesman problem, and visuomotor control of robots. |
 Principal component
analysis and long time protein dynamics. Manel A. Balsera, Willy Wriggers,
Yoshitsugu Oono, and Klaus Schulten. Journal of Physical Chemistry,
100:2567-2572, 1996.
Principal component
analysis and long time protein dynamics. Manel A. Balsera, Willy Wriggers,
Yoshitsugu Oono, and Klaus Schulten. Journal of Physical Chemistry,
100:2567-2572, 1996. (Citations: 306) It has been suggested that principal component analysis can identify slow modes in proteins and, thereby, facilitate the study of long time dynamics. However, sampling errors due to finite simulation times preclude the identification of slow modes that can be used for this purpose. This is demonstrated numerically with the aid of G-actin simulations, and analytically with the aid of a model which is exactly recoverable by Principal Component Analysis. |
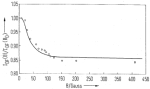 Magnetic field dependence of the geminate recombination of radical ion pairs in polar solvents. Klaus Schulten, H. Staerk,
Albert Weller, Hans-Joachim Werner, and B. Nickel. Zeitschrift
für Physikalische Chemie, NF101:371-390, 1976.
Magnetic field dependence of the geminate recombination of radical ion pairs in polar solvents. Klaus Schulten, H. Staerk,
Albert Weller, Hans-Joachim Werner, and B. Nickel. Zeitschrift
für Physikalische Chemie, NF101:371-390, 1976. (Citations: 299) Pairs of radical ions are generated in polar solvents by nanosecond laser flashes in a singlet electron spin state via photoinduced electron transfer. The recombination monitored spectroscopically with a time resolution |
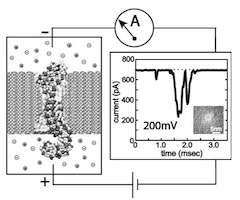 Microscopic kinetics of DNA translocation through synthetic nanopore. Aleksij Aksimentiev, Jiunn Benjamin Heng, Gregory Timp, and Klaus Schulten. Biophysical Journal, 87:2086-2097, 2004
Microscopic kinetics of DNA translocation through synthetic nanopore. Aleksij Aksimentiev, Jiunn Benjamin Heng, Gregory Timp, and Klaus Schulten. Biophysical Journal, 87:2086-2097, 2004 (Citations: 295) We have previously demonstrated that a nanometer-diameter pore in. nanometer-thick MOS (Metal-Oxide-Semiconductor)-compatible membrane can be used as a molecular sensor for detecting DNA. The prospects for using this type of mechanism for sequencing DNA are avidly being pursued. The key attribute of the sensor is the electric field-induced (voltage-driven) translocation of the DNA molecule in an electrolytic solution across the membrane through the nanopore. To complement ongoing experimental studies developing such pores and measuring signals in response to the presence of DNA, we conducted molecular dynamics simulations of DNA translocation through the nanopore. A typical simulated system included a patch of a silicon nitride membrane dividing water solution of potassium chloride into two compartments connected by the nanopore. External electrical fields induced capturing of the DNA molecules by the pore from the solution and subsequent translocation. Molecular dynamics simulations suggest that 20 base pairs double stranded DNA can transit a nanopore of. 2.4 |
 Ten-microsecond molecular dynamics simulation of a fast-folding WW domain.
Peter L. Freddolino, Feng Liu, Martin Gruebele, and Klaus Schulten.
Biophysical Journal, 94:L75-L77, 2008.
Ten-microsecond molecular dynamics simulation of a fast-folding WW domain.
Peter L. Freddolino, Feng Liu, Martin Gruebele, and Klaus Schulten.
Biophysical Journal, 94:L75-L77, 2008. (Citations: 291) All-atom molecular dynamics (MD) simulations of protein folding allow analysis of the folding process at an unprecedented level of detail. Unfortunately, such simulations have not yet reached their full potential both due to difficulties in sufficiently sampling the microsecond timescales needed for folding, and because the force field used may yield neither the correct dynamical sequence of events nor the folded structure. The ongoing study of protein folding through computational methods thus requires both improvements in the performance of molecular dynamics programs to make longer timescales accessible, and testing of force fields in the context of folding simulations. We report a ten-microsecond simulation of an incipient downhill-folding WW domain mutant along with measurement of a molecular time and activated folding time of 1.5 microseconds and 13.3 microseconds, respectively. The protein simulated in explicit solvent exhibits several metastable states with incorrect topology and does not assume the native state during the present simulations. |
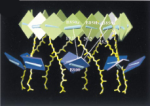 Pigment organization
and transfer of electronic excitation in the purple bacteria. Xiche Hu,
Thorsten Ritz, Ana Damjanovic, and Klaus Schulten. Journal of Physical
Chemistry B, 101:3854-3871, 1997.
Pigment organization
and transfer of electronic excitation in the purple bacteria. Xiche Hu,
Thorsten Ritz, Ana Damjanovic, and Klaus Schulten. Journal of Physical
Chemistry B, 101:3854-3871, 1997. (Citations: 289) Absorption of light by light harvesting complexes and transfer of electronic excitation to the photosynthetic reaction center (RC) constitutes the primary light harvesting process of photosynthesis. This process is investigated on the basis of an atomic level structure of the so-called photosynthetic unit of the photosynthetic bacterium Rb. sphaeroides. The photosynthetic unit combines in the intracytoplasmic membrane a nanometric assembly of three protein complexes: (i) the photosynthetic reaction center, (ii) a ring-shaped light harvesting complex LH-I, and (iii) multiple copies of a similar complex, LH-II. The unit has been modeled using the known structure of (i) and for (ii) a model, recently obtained and complexed appropriately with (i); for (iii) the structure of LH-II of Rs. molischianum is substituted. The model describes in detail the organization of chromophores involved in primary light absorption and excitation transfer: a hierarchy of ring-shaped chlorophyll aggregates which surround four centrally located chlorophylls of the photosynthetic reaction center. The chlorophylls involved in the overall transfer are found in a co-planar arrangements. On the basis of the modeled structure a quantum-mechanical description of the entire light harvesting process is developed. For this purpose an effective Hamiltonian is established a priori and then employed to describe the LH-II |
 Three-dimensional neural net for learning visuo-motor coordination of a robot arm.
Thomas Martinetz, Helge Ritter, and Klaus Schulten.
IEEE Transactions on Neural Networks, 1:131-136, 1990.
Three-dimensional neural net for learning visuo-motor coordination of a robot arm.
Thomas Martinetz, Helge Ritter, and Klaus Schulten.
IEEE Transactions on Neural Networks, 1:131-136, 1990. (Citations: 289) An extension of Kohonen's self-organizing mapping algorithm together with an error-correction scheme based on the Widrow-Hoff learning rule is applied to develop a learning algorithm for the visuomotor coordination of a simulated robot arm. Learning occurs by a sequence of trial movements without the need of an external teacher. Using input signals from a pair of cameras, the"closed" robot arm system is able to reduce its positioning error to about 0.3 percent of the linear dimensions of its work space. This is achieved by choosing the connectivity of a 3D-lattice between the units of the neural net. |
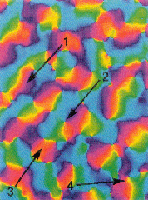 Models of orientation
and ocular dominance columns in the visual cortex: A critical comparison.
Edgar Erwin, Klaus Obermayer, and Klaus Schulten. Neural Computation,
7:425-468, 1995.
Models of orientation
and ocular dominance columns in the visual cortex: A critical comparison.
Edgar Erwin, Klaus Obermayer, and Klaus Schulten. Neural Computation,
7:425-468, 1995. (Citations: 286) Orientation and ocular dominance maps in the primary visual cortex of mammals are among the most thoroughly investigated of the patterns in the cerebral cortex. A considerable amount of work has been dedicated to unraveling both their detailed structure and the neural mechanisms that underlie their formation and development. Many schemes have been proposed, some of which are in competition. Some models focus on development of receptive fields while others focus on the structure of cortical maps, i.e., the arrangement of receptive field properties across the cortex. Each model used different means to determine its success at reproducing experimental map patterns, often relying principally on visual comparison. Experimental data are becoming available that allow a more careful evaluation of models. In this contribution more than 10 of the most prominent models of cortical map formation and structure are critically evaluated and compared with the most recent experimental findings from macaque striate cortex. Comparisons are based on properties of the predicted or measured cortical map patterns. We introduce several new measures for comparing experimental and model map data that reveal important differences between models. We expect that the use of these measures will improve current models by helping determine parameters to match model maps to experimental data now becoming available from a variety of species. Our study reveals that (1) despite apparent differences, many models are based on similar principles and consequently make similar predictions, (2) several models produce orientation map patterns that are not consistent with the experimental data from macaques, regardless of the plausibility of the models' suggested physiological implementations, and (3) no models have yet fully accounted for both the local and the global relationships between orientation and ocular dominance map patterns. |
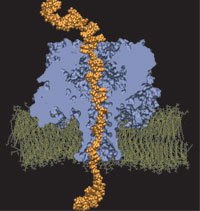 Orientation
discrimination of single stranded DNA inside the α-hemolysin membrane
channel. Jerome Mathé, Aleksei Aksimentiev, David R. Nelson,
Klaus Schulten, and Amit Meller. Proceedings of the National Academy of
Sciences, USA, 102:12377-12382, 2005 Orientation
discrimination of single stranded DNA inside the α-hemolysin membrane
channel. Jerome Mathé, Aleksei Aksimentiev, David R. Nelson,
Klaus Schulten, and Amit Meller. Proceedings of the National Academy of
Sciences, USA, 102:12377-12382, 2005 (Citations: 283) We characterize the voltage-driven and voltage-free motion of single stranded DNA (ssDNA) molecules captured inside the |
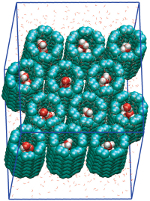 Water and proton conduction through carbon nanotubes as models for biological channels. Fangqiang Zhu and Klaus Schulten. Biophysical Journal, 85:236-244, 2003 Water and proton conduction through carbon nanotubes as models for biological channels. Fangqiang Zhu and Klaus Schulten. Biophysical Journal, 85:236-244, 2003 (Citations: 280) Carbon nanotubes, unmodified (pristine) and modified through charged atoms, were simulated in water, and their water conduction rates determined. The conducted water inside the nanotubes was found to exhibit a strong ordering of its dipole moments. In pristine nanotubes the water dipoles adopt a single orientation along the tube axis with a low flipping rate between the two possible alignments. Modification can induce in nanotubes a bipolar ordering as previously observed in biological water channels. Network thermodynamics was applied to investigate proton conduction through the nanotubes. |
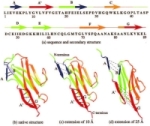 The key event in force-induced unfolding of titin's immunoglobulin domains. Hui Lu and Klaus Schulten. Biophysical Journal,
79:51-65, 2000.
The key event in force-induced unfolding of titin's immunoglobulin domains. Hui Lu and Klaus Schulten. Biophysical Journal,
79:51-65, 2000. (Citations: 267) Steered molecular dynamics simulation of force-induced titin immunoglobulin domain I27 unfolding led to the discovery of a significant potential energy barrier at an extension of about 14 Å on the unfolding pathway that protects the domain against stretching. Previous simulations showed that this barrier is due to the concurrent breaking of six interstrand hydrogen bonds (H-bonds) between |
 Lipid bilayer pressure profiles and mechanosensitive channel gating. Justin Gullingsrud and Klaus Schulten. Biophysical Journal, 86:3496-3509, 2004 Lipid bilayer pressure profiles and mechanosensitive channel gating. Justin Gullingsrud and Klaus Schulten. Biophysical Journal, 86:3496-3509, 2004 (Citations: 262) The function of membrane proteins often depends on the proteins interaction with their lipid environment, spectacularly so in the case of mechanosensitive channels, which are gated through tension mediated by the surrounding lipids. Lipid bilayer tension is distributed quite inhomogeneously, but neither the scale at which relevant variation takes place nor the effect of varying lipid composition or tension have yet been investigated in atomic detail. We calculated lateral pressure profile distributions in lipid bilayers of various composition from all-atom molecular dynamics simulations totaling 110.5 ns in length. Reproducible pressure profile features at the. |
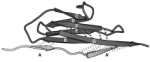 Forced unfolding of the fibronectin type III module reveals a tensile molecular recognition switch. André Krammer,
Hui Lu, Barry Isralewitz, Klaus Schulten, and Viola Vogel. Proceedings of the
National Academy of Sciences, USA, 96:1351-1356, 1999.
Forced unfolding of the fibronectin type III module reveals a tensile molecular recognition switch. André Krammer,
Hui Lu, Barry Isralewitz, Klaus Schulten, and Viola Vogel. Proceedings of the
National Academy of Sciences, USA, 96:1351-1356, 1999. (Citations: 255) The tenth type III module of fibronectin, |
 A principle for the
formation of the spatial structure of cortical feature maps. Klaus
Obermayer, Helge Ritter, and Klaus Schulten. Proceedings of the National
Academy of Sciences, USA, 87:8345-8349, 1990.
A principle for the
formation of the spatial structure of cortical feature maps. Klaus
Obermayer, Helge Ritter, and Klaus Schulten. Proceedings of the National
Academy of Sciences, USA, 87:8345-8349, 1990. (Citations: 255) Orientation-selective cells in the striate cortex of higher animals are organized as a hierarchical topographic map of two stimulus features: (i) position in visual space and (ii) orientation. We show that the observed structure of the topographic map can arise from a principle of continuous mapping. For the realization of this principle we use a mathematical model that can be interpreted as an adaptive process changing a set of synaptic weights, or synaptic connection strengths, between two layers of cells. The patterns of orientation preference and selectivity generated by the model are similar to the patterns seen in the visual cortex of macaque monkey and cat and correspond to a neural projection that maps a more than two-dimensional feature space onto a two-dimensional cortical surface under the constraint that shape and position of the receptive fields of the neurons vary smoothly over the cortical surface. |
 Steered molecular dynamics investigations of protein function.
Barry Isralewitz, Jerome Baudry, Justin Gullingsrud, Dorina Kosztin, and Klaus Schulten.
Journal of Molecular Graphics and Modeling, 19:13-25, 2001.
Steered molecular dynamics investigations of protein function.
Barry Isralewitz, Jerome Baudry, Justin Gullingsrud, Dorina Kosztin, and Klaus Schulten.
Journal of Molecular Graphics and Modeling, 19:13-25, 2001. (Citations: 252) Molecular recognition and mechanical properties of proteins govern molecular processes in the cell that can cause disease and can be targeted for drug design. Single molecule measurement techniques have greatly advanced knowledge but cannot resolve enough detail to be interpreted in terms of protein structure. We seek to complement, therefore, the observations through so-called Steered Molecular Dynamics (SMD) simulations that on the one hand link directly to experiments and and the other hand provide atomic level descriptions of the underlying events. Such a research program has been initiated in our group and has involved, for example, studies of elastic properties of immunoglobulin and fibronectin domains as well as the binding of biotin and avidin. In this article, we explain the SMD method and suggest how it can be applied to the function of three systems that are the focus of modern molecular biology research, force transduction by the muscle protein titin and extracellular matrix protein fibronectin, recognition of antibody-antigene pairs, and ion selective conductivity of the K+ channel. |
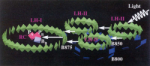 How nature harvests
sunlight. Xiche Hu and Klaus Schulten. Physics Today, 50:28-34, 1997.
How nature harvests
sunlight. Xiche Hu and Klaus Schulten. Physics Today, 50:28-34, 1997. (Citations: 248) Specialized molecular aggregates in purple bacteria exploit subtle quantum physics to collect and convert light energy for photosynthesis. |
 Imaging the migration pathways for O2, CO, NO, and Xe inside myoglobin.
Jordi Cohen, Anton Arkhipov, Rosemary Braun, and Klaus Schulten.
Biophysical Journal, 91:1844-1857, 2006.
Imaging the migration pathways for O2, CO, NO, and Xe inside myoglobin.
Jordi Cohen, Anton Arkhipov, Rosemary Braun, and Klaus Schulten.
Biophysical Journal, 91:1844-1857, 2006. (Citations: 242) Myoglobin (Mb) is perhaps the most studied protein, experimentally and theoretically. Despite the wealth of known details regarding the gas migration processes inside Mb, there exists no fully conclusive picture of these pathways. We address this deficiency by presenting a complete map of all the gas migration pathways inside Mb for small gas ligands (O2, NO, CO, and Xe). To accomplish this, we introduce a computational approach for studying gas migration, which we call implicit ligand sampling. Rather than simulating actual gas migration events, we infer the location of gas migration pathways based on a free-energy perturbation approach applied to simulations of Mb's dynamical fluctuations at equilibrium in the absence of ligand. The method provides complete three-dimensional maps of the potential of mean force of gas ligand placement anywhere inside a protein-solvent system. From such free-energy maps we identify each gas docking site, the pathways between these sites, to the heme and to the external solution. Our maps match previously known features of these pathways in Mb, but also point to the existence of additional exits from the protein matrix in regions that are not easily probed by experiment. We also compare the pathway maps of Mb for different gas ligands and for different animal species. |
 Coarse grained protein-lipid model with application to lipoprotein particles.
Amy Y. Shih, Anton Arkhipov, Peter L. Freddolino, and Klaus Schulten.
Journal of Physical Chemistry B, 110:3674-3684, 2006.
Coarse grained protein-lipid model with application to lipoprotein particles.
Amy Y. Shih, Anton Arkhipov, Peter L. Freddolino, and Klaus Schulten.
Journal of Physical Chemistry B, 110:3674-3684, 2006. (Citations: 239) A coarse-grained model for molecular dynamics simulations is extended from lipids to proteins. In the framework of such models pioneered by Klein, atoms are described group-wise by beads, with the interactions between beads governed by effective potentials. The extension developed here is based on a coarse-grained lipid model previously developed by Marrink et al., though further versions will reconcile the approach taken with the systematic approach of Klein and other authors. Each amino acid of the protein is represented by two coarse-grained beads, one for the backbone (identical for all residues) and one for the side-chain (which differs depending on the residue type). The coarse-graining reduces system size about ten-fold and allows integration time steps of 25 to 50 fs. The model is applied to simulations of discoidal high-density lipoprotein particles, involving water, lipids, and two primarily helical proteins. These particles are an ideal test system for the extension of coarse-grained models. Our model proved reliable in maintaining the shape of pre-assembled particles and in accurately reproducing overall structural features of high-density lipoproteins. Microsecond simulations of lipoprotein assembly revealed formation of a protein-lipid complex in which two proteins are attached to either side of a discoidal lipid bilayer. |
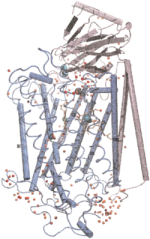 Oxygen and proton
pathways in cytochrome c oxidase. Ivo Hofacker and Klaus Schulten.
PROTEINS: Structure, Function, and Genetics, 30:100-107, 1998.
Oxygen and proton
pathways in cytochrome c oxidase. Ivo Hofacker and Klaus Schulten.
PROTEINS: Structure, Function, and Genetics, 30:100-107, 1998. (Citations: 239) Background: Cytochrome c oxidase is a redox-driven proton pump, which couples the reduction of oxygen to water to the translocation of protons across the membrane. The recently solved x-ray structures of cytochrome c oxidase permit molecular dynamics simulations of the underlying transport processes. To eventually establish the proton pump mechanism we investigate the transport of the substrates, oxygen and protons, through the enzyme. Results: Molecular dynamics simulations of oxygen diffusion through the protein reveal a pathway to the oxygen binding site starting at a hydrophobic cavity near the membrane exposed surface of subunit I, close to the interface to subunit III. A large number of water sites is predicted within the protein. The water molecules form two channels along which protons can enter from the cytoplasmic (matrix) side of the protein and reach the binuclear center. Conclusions: Oxygen is channeled to the catalytic center of the enzyme along a well defined path. Hydrophobic cavities at the start of the path could serve as reservoirs for oxygen. Water might play an essential role for the transfer of protons in cytochrome c oxidase. A possible pumping mechanism is proposed that involves a shuttling motion of a glutamic acid side chain, which could then transfer a proton to a propionate group of heme |
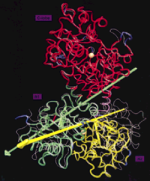 Protein domain movements: Detection of rigid domains
and visualization of hinges in comparisons of atomic coordinates. Willy Wriggers and Klaus Schulten.
PROTEINS: Structure, Function, and Genetics, 29:1-14, 1997.
Protein domain movements: Detection of rigid domains
and visualization of hinges in comparisons of atomic coordinates. Willy Wriggers and Klaus Schulten.
PROTEINS: Structure, Function, and Genetics, 29:1-14, 1997.(Citations: 237) The activity of many proteins induces conformational transitions by hinge-bending, which involves the movement of relatively rigid parts of a protein about flexible joints. We present an algorithm to identify and visualize the movements of rigid domains about common hinges in proteins. In comparing two structures, the method partitions a protein into domains of preserved geometry. The domains are extracted by an adaptive selection procedure using least-squares fitting. The user can maintain the spatial connectivity of the domains and filter significant structural differences (domain movements) from noise in the compared sets of atomic coordinates. The algorithm subsequently characterizes the relative movements of the found domains by effective rotation-axes (hinges). The method is applied to several known instances of domain movements in protein structures, namely, in lactoferrin, hexokinase, actin, the extracellular domains of human tissue factor, and of the receptor of human growth factor. The results are visualized with the molecular graphics package VMD (Humphrey et al., J. Mol. Graphics 14(1):33-38, 1996). Applications of the algorithm to the analysis of conformational changes in proteins and to biomolecular docking are discussed. |
 Theory and simulation of water permeation in aquaporin-1.
Fangqiang Zhu, Emad Tajkhorshid, and Klaus Schulten.
Biophysical Journal, 86:50-57, 2004.
Theory and simulation of water permeation in aquaporin-1.
Fangqiang Zhu, Emad Tajkhorshid, and Klaus Schulten.
Biophysical Journal, 86:50-57, 2004. (Citations: 232) The paper starts with a detailed discussion of the difference between osmotic permeability |
 Excitons in a photosynthetic light-harvesting system: A combined
molecular dynamics, quantum chemistry and polaron model study.
Ana Damjanovic, Ioan Kosztin, Ulrich Kleinekathoefer, and Klaus Schulten.
Physical Review E, 65:031919, 2002.
Excitons in a photosynthetic light-harvesting system: A combined
molecular dynamics, quantum chemistry and polaron model study.
Ana Damjanovic, Ioan Kosztin, Ulrich Kleinekathoefer, and Klaus Schulten.
Physical Review E, 65:031919, 2002. (Citations: 232) The dynamics of pigment-pigment and pigment-protein interactions in light-harvesting complexes is studied with a novel approach that combines molecular dynamics simulations with quantum chemistry calculations and a polaron model analysis. The molecular dynamics simulation of light-harvesting complexes was performed on an 87,055 atom system comprised of an LH-II complex of Rhodospirillum molischianum embedded in a lipid bilayer and surrounded with appropriate water layers. For each of the 16 B850 BChls we performed 400 ab initio quantum chemistry calculations on geometries that emerged from the molecular dynamical simulations, determining the fluctuations of pigment excitation energies as a function of time. From the results of these calculations we construct a time-dependent Hamiltonian of the B850 exciton system from which we determine within linear response theory the absorption spectrum. Finally, a polaron model is introduced to describe both the excitonic and coupled phonon degrees of freedom by quantum mechanics. The exciton-phonon coupling that enters into the polaron model, and the corresponding phonon spectral function are derived from the molecular dynamics and quantum chemistry simulations. The model predicts that excitons in the B850 BChl ring are delocalized over five pigments at room temperature. Also, the polaron model permits the calculation of the absorption and circular dichroism spectra of the B850 excitons from the sole knowledge of the autocorrelation function of the excitation energies of individual BChls, which is readily available from the combined molecular dynamics and quantum chemistry simulations. The obtained results are found to be in good agreement with the experimentally measured absorption and circular dichroism spectra. |
 In search of the hair-cell gating spring: Elastic properties of ankyrin and cadherin repeats.
Marcos Sotomayor, David P. Corey, and Klaus Schulten.
Structure, 13:669-682, 2005.
In search of the hair-cell gating spring: Elastic properties of ankyrin and cadherin repeats.
Marcos Sotomayor, David P. Corey, and Klaus Schulten.
Structure, 13:669-682, 2005. (Citations: 230) Mechanotransduction in vertebrate hair cells involves a biophysically defined elastic element (the ``gating spring'') that pulls on the transduction channels. The tip link, a fine filament made of cadherin 23 linking adjacent stereocilia in hair-cell bundles, has been suggested to be the gating spring. However, TRP channels that mediate mechanotransduction in Drosophila, zebrafish, and mice often have cytoplasmic domains containing a large number of ankyrin repeats that are also candidates for the gating spring. We have explored the elastic properties of cadherin and ankyrin repeats through molecular dynamics simulations using crystallographic structures of proteins with one cadherin repeat or 4 and 12 ankyrin repeats, and using models of 17 and 24 ankyrin repeats. The extension and stiffness of large ankyrin-repeat structures were found to match those predicted by the gating-spring model. Our results suggest that ankyrin repeats of TRPA1 and TRPN1 channels serve as the gating spring for mechanotransduction. |
 Statistical-mechanical
analysis of self-organization and pattern formation during the development of
visual maps. Klaus Obermayer, Gary G. Blasdel, and Klaus Schulten.
Physical Review A, 45:7568-7589, 1992.
Statistical-mechanical
analysis of self-organization and pattern formation during the development of
visual maps. Klaus Obermayer, Gary G. Blasdel, and Klaus Schulten.
Physical Review A, 45:7568-7589, 1992. (Citations: 230) We report a detailed analytical and numerical model study of pattern formation during the development of visual maps, namely, the formation of topographic maps and orientation and ocular dominance columns in the striate cortex. Pattern formation is described by a stimulus-driven Markovian process, the self-organizing feature map. This algorithm generates topologically correct maps between a space of (visual) input signals and an array of formal "neurons," which in our model represents the cortex. We define order parameters that are a function of the set of visual stimuli an animal perceives, and we demonstrate that the formation of orientation and ocular dominance columns is the result of a global instability of the retinoptic projection above a critical value of these order parameters. We characterize the spatial structure of the emerging patterns by power spectra, correlation functions, and Gabor transforms, and we compare model predictions with experimental data obtained from the striate cortex of the macaque monkey with optical imaging. Above the critical value of the order parameters the model predicts a lateral segregation of the striate cortex into (i) binocular regions with linear changes in orientation preference, where iso-orientation slabs run perpendicular to the ocular dominance bands, and (ii) monocular regions with low orientation specificity, which contain the singularities of the orientation map. Some of these predictions have already been verified by experiments. |
 Pressure-induced water transport in membrane channels studied by molecular dynamics.
Fangqiang Zhu, Emad Tajkhorshid, and Klaus Schulten.
Biophysical Journal, 83:154-160, 2002.
Pressure-induced water transport in membrane channels studied by molecular dynamics.
Fangqiang Zhu, Emad Tajkhorshid, and Klaus Schulten.
Biophysical Journal, 83:154-160, 2002. (Citations: 224) A method is proposed to measure the water permeability of membrane channels by means of molecular dynamics simulations. By applying a constant force to the bulk water molecules and a counter force on the complementary system, a hydrostatic pressure difference across the membrane can be established, producing a net directional water flow. The hydraulic or osmotic permeability can then be determined by the ratio of the water flux and the pressure difference. The method is applied and tested on an aquaglyceroporin channel through a series of simulations totaling 5 ns in duration. |
 A mechanism for the
light-driven proton pump of Halobacterium halobium. Klaus Schulten and
Paul Tavan. Nature, 272:85-86, 1978.
A mechanism for the
light-driven proton pump of Halobacterium halobium. Klaus Schulten and
Paul Tavan. Nature, 272:85-86, 1978. (Citations: 221) Mitchell's hypothesis of chemiosmotic coupling between redox reactions and ATP synthesis in membranes is supported by the finding of a light-driven proton pump in the purple membrane of Halobacterium halobium. The purple membrane contains the protein bacteriorhodopsin in a crystalline array, with retinal as a chromophore. We propose here, on the basis of quantumchemical arguments and experimental observations, that the H. halobium proton pump may involve proton translocation through photoisomerisation of retinal about its 14-15 single bond. |
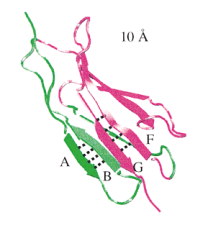 Steered molecular
dynamics simulations of force-induced protein domain unfolding. Hui Lu and
Klaus Schulten. PROTEINS: Structure, Function, and Genetics, 35:453-463, 1999.
Steered molecular
dynamics simulations of force-induced protein domain unfolding. Hui Lu and
Klaus Schulten. PROTEINS: Structure, Function, and Genetics, 35:453-463, 1999.
(Citations: 219) Steered molecular dynamics (SMD), a computer simulation method for studying force-induced reactions in biopolymers, has been applied to investigate the response of protein domains to stretching apart of their terminal ends. The simulations mimic atomic force microscopy and optical tweezer experiments, but proceed on much shorter time scales. The simulations on different domains for 0.6 nanosecond each reveal two types of protein responses: the first type, arising in certain |
 Algorithmic
challenges in computational molecular biophysics. Tamar Schlick,
Robert Skeel, Axel Brünger, Laxmikant Kalé, John A. Board Jr., Jan Hermans, and Klaus Schulten. Journal of Computational Physics, 151:9-48, 1999 Algorithmic
challenges in computational molecular biophysics. Tamar Schlick,
Robert Skeel, Axel Brünger, Laxmikant Kalé, John A. Board Jr., Jan Hermans, and Klaus Schulten. Journal of Computational Physics, 151:9-48, 1999 (Citations: 217) A perspective of biomolecular simulations today is given, with illustrative applications and an emphasis on algorithmic challenges, as reflected by the work of a multidisciplinary team of investigators from five institutions. Included are overviews and recent descriptions of algorithmic work in long-time integration for molecular dynamics, fast electrostatic evaluations, crystallographic refinement approaches, and implementaiton of large, computer-intensive programs on modern architectures. |
 Steered molecular dynamics. Sergei Izrailev, Sergey Stepaniants, Barry Isralewitz, Dorina Kosztin, Hui Lu, Ferenc Molnar, Willy Wriggers, and Klaus Schulten. In P. Deuflhard, J. Hermans, B. Leimkuhler, A. E. Mark, S. Reich, and
R. D. Skeel, editors, Computational Molecular Dynamics: Challenges,
Methods, Ideas, volume 4 of Lecture Notes in Computational Science and
Engineering, pp. 39-65. Springer-Verlag, Berlin, 1998.
Steered molecular dynamics. Sergei Izrailev, Sergey Stepaniants, Barry Isralewitz, Dorina Kosztin, Hui Lu, Ferenc Molnar, Willy Wriggers, and Klaus Schulten. In P. Deuflhard, J. Hermans, B. Leimkuhler, A. E. Mark, S. Reich, and
R. D. Skeel, editors, Computational Molecular Dynamics: Challenges,
Methods, Ideas, volume 4 of Lecture Notes in Computational Science and
Engineering, pp. 39-65. Springer-Verlag, Berlin, 1998. (Citations: 206) Steered molecular dynamics (SMD) induces unbinding of ligands and conformational changes in biomolecules on time scales accessible to molecular dynamics simulations. Time-dependent external forces are applied to a system, and the responses of the system are analyzed. SMD has already provided important qualitative insights into biologically relevant problems, as demonstrated here for applications ranging from identification of ligand binding pathways to explanation of elastic properties of proteins. First attempts to deduce potentials of mean force by discounting irreversible work performed on the system are summarized. The non-equilibrium statistical mechanics underlying analysis of SMD data is outlined. |
 Accelerated molecular dynamics simulation with the parallel fast multipole algorithm. John A. Board, Jr., J. W. Causey, James
F. Leathrum, Jr., Andreas Windemuth, and Klaus Schulten.
Chemical Physics Letters, 198:89-94, 1992.
Accelerated molecular dynamics simulation with the parallel fast multipole algorithm. John A. Board, Jr., J. W. Causey, James
F. Leathrum, Jr., Andreas Windemuth, and Klaus Schulten.
Chemical Physics Letters, 198:89-94, 1992. (Citations: 202) We have implemented the fast multipole algorithm (FMA) of Greengard and Rokhlin and incorporated it into the molecular dynamics program MD of Windemuth and Schulten, allowing rapid computation of the non-bonded forces acting in dynamical protein systems without truncation or other corruption of the Coulomb force. The resulting program speeds up simulations of protein systems with approximately 24000 atoms by up to an order of magnitude on a single workstation. Additionally, we have implemented a parallel version of the three-dimensional FMA code on a loosely coupled network of workstations, further reducing simulation times. Large (in both size of system and length of simulated time) protein molecular dynamics simulations are now possible on workstations rather than supercomputers, and very large protein computations are possible on clusters of workstations and parallel machines. |
 The electromechanics of DNA in a synthetic nanopore.
J. B. Heng, A. Aksimentiev, C. Ho, P. Marks, Y. V. Grinkova, S. Sligar, K. Schulten, and G. Timp.
Biophysical Journal, 90:1098-1106, 2006.
The electromechanics of DNA in a synthetic nanopore.
J. B. Heng, A. Aksimentiev, C. Ho, P. Marks, Y. V. Grinkova, S. Sligar, K. Schulten, and G. Timp.
Biophysical Journal, 90:1098-1106, 2006. (Citations: 196) We have explored the electromechanical properties of DNA on a nanometer-length scale using an electric field to force single molecules through synthetic nanopores in ultra-thin silicon nitride membranes. At low electric fields E <200mV/10nm, we observed that single stranded DNA can permeate pores with a diameter |
 Development of
feature detectors by self-organization: A network model. Jeanne Rubner and Klaus Schulten. Biological Cybernetics, 62:193-199, 1990 Development of
feature detectors by self-organization: A network model. Jeanne Rubner and Klaus Schulten. Biological Cybernetics, 62:193-199, 1990 (Citations: 184) We present a two-layered network of linear neurons that organizes itself as to extract the complete information contained in a set of presented patterns. The weights between layers obey. Hebbian rule. We propose a local anti-Hebbian rule for lateral, hierarchically organized weights within the output layer. This rule forces the activities of the output units to become uncorrelated and the lateral weights to vanish. The weights between layers converge to the eigenvectors of the covariance matrix of input patterns, i.e., the network performs a principal component analysis, yielding all principal components. As a consequence of the proposed learning scheme, the output units become detectors of orthogonal features, similar to ones found in the brain of mammals. |
 Kohonen's self-organizing maps: Exploring their computational capabilities.
Helge Ritter and Klaus Schulten.
In IEEE International Conference on Neural Networks, San Diego,
California, July 24-27, 1988, volume 1, pp. 109-116, New York, 1988. The
Institute of Electrical and Electronics Engineers.
Kohonen's self-organizing maps: Exploring their computational capabilities.
Helge Ritter and Klaus Schulten.
In IEEE International Conference on Neural Networks, San Diego,
California, July 24-27, 1988, volume 1, pp. 109-116, New York, 1988. The
Institute of Electrical and Electronics Engineers. (Citations: 179) It is demonstrated that the computational capabilities of Kohonen's self-organizing mapping algorithm can be applied to problems from such diverse fields as sensory mappings, combinatorial optimization and motor control. In addition we present some recent mathematical results characterizing important properties of the algorithm in these situations. |
 Stretching DNA using an electric field in a synthetic nanopore.
J. B Heng, A. Aksimentiev, C. Ho, P. Marks, Y. V. Grinkova, S. Sligar, K. Schulten, and G. Timp.
Nano Letters, 5:1883-1888, 2005.
Stretching DNA using an electric field in a synthetic nanopore.
J. B Heng, A. Aksimentiev, C. Ho, P. Marks, Y. V. Grinkova, S. Sligar, K. Schulten, and G. Timp.
Nano Letters, 5:1883-1888, 2005. (Citations: 177) The mechanical properties of DNA over segments comparable to the size of a protein-binding site (3-10nm) are examined using an electric field-induced translocation of single molecules through a nanometer diameter pore. DNA, immersed in an electrolyte, is forced through synthetic pores ranging from 0.5 to 1.5nm in radius in a 10nm thick Si |
 Molecular dynamics investigation of primary photoinduced events in the activation of rhodopsin. Jan Saam, Emad Tajkhorshid, Shigehiko Hayashi, and Klaus Schulten. Biophysical Journal, 83:3097-3112, 2002
Molecular dynamics investigation of primary photoinduced events in the activation of rhodopsin. Jan Saam, Emad Tajkhorshid, Shigehiko Hayashi, and Klaus Schulten. Biophysical Journal, 83:3097-3112, 2002 (Citations: 167) Retinal cis-trans isomerization and early relaxation steps have been studied in a 10ns molecular dynamics simulation of a fully hydrated model of membrane-embedded rhodopsin. The isomerization, induced by transiently switching the potential energy function governing the C |
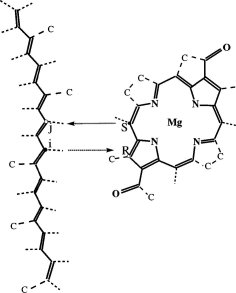 Energy transfer between carotenoids and bacteriochlorophylls in light-harvesting complex II of purple bacteria. Ana Damjanovic, Thorsten Ritz, and Klaus Schulten. Physical Review E, 59:3293-3311, 1999 Energy transfer between carotenoids and bacteriochlorophylls in light-harvesting complex II of purple bacteria. Ana Damjanovic, Thorsten Ritz, and Klaus Schulten. Physical Review E, 59:3293-3311, 1999 (Citations: 169) In photosynthetic light harvesting systems carotenoids and chlorophylls jointly absorb light and transform its energy within about a picosecond into electronic singlet excitations of the chlorophylls only. This paper investigates this process for the light harvesting complex II of the purple bacterium Rs. molischianum, for which a structure and, hence, the exact arrangement of the participating chlorophylls and carotenoids have recently become known. Based on this structure and on CI expansions of the electronic states of individual chromophores (chlorophylls and carotenoids) as well as on an exciton description of a circular aggregate of chlorophylls, the excitation transfer between carotenoids and chlorophylls is described by means of Fermi's golden rule. The electronic coupling between the various electronic excitations is determined for all orders of multipoles (generalized Förster mechanism) and includes the electron exchange (dexter mechanism) term. The rates and efficiencies for different pathways of excitation transfer, e.g., |
 Self-organizing maps: Stationary states, metastability and convergence rate.
Edgar Erwin, Klaus Obermayer, and Klaus Schulten.
Biological Cybernetics, 67:35-45, 1992.
Self-organizing maps: Stationary states, metastability and convergence rate.
Edgar Erwin, Klaus Obermayer, and Klaus Schulten.
Biological Cybernetics, 67:35-45, 1992. (Citations: 168) We investigate the effect of various types of neighborhood function on the convergence rates and the presence or absence of metastable stationary states of Kohonen's self-organizing feature map algorithm in one dimension. We demonstrate that the time necessary to form a topographic representation of the unit interval [0,1] may vary over several orders of magnitude depending on the range and also the shape of the neighborhood function, by which the weight changes of the neurons in the neighborhood of the winning neuron are scaled. We will prove that for neighborhood functions which are convex on an interval given by the length of the Kohonen chain there exist no metastable states. For all other neighborhood functions, metastable states are present and may trap the algorithm during the learning process. For the widely-used Gaussian function there exists a threshold for the width above which metastable states cannot exist. Due to the presence or absence of metastable states, convergence time is very sensitive to slight changes in the shape of the neighborhood function. Fastest convergence is achieved using neighborhood functions which are "convex" over a large range around the winner neuron and yet have large differences in value at neighboring neurons. |
 Structure and functional significance of mechanically unfolded
fibronectin type III1 intermediates.
Mu Gao, David Craig, Olivier Lequin, Iain D. Campbell, Viola Vogel, and Klaus Schulten.
Proceedings of the National Academy of Sciences, USA, 100:14784-14789,
2003.
Structure and functional significance of mechanically unfolded
fibronectin type III1 intermediates.
Mu Gao, David Craig, Olivier Lequin, Iain D. Campbell, Viola Vogel, and Klaus Schulten.
Proceedings of the National Academy of Sciences, USA, 100:14784-14789,
2003. (Citations: 167) Fibronectin (FN) forms fibrillar networks coupling cells to the extracellular matrix (ECM). The formation of FN fibrils, fibrillogenesis, is a tightly regulated process involving the exposure of cryptic binding sites in individual FN type III (FN-III) repeats presumably exposed by mechanical tension. The FN-III1 module has been previously proposed to contain such cryptic sites that promote the assembly of extracellular matrix FN fibrils. We have combined NMR and steered molecular dynamics (SMD) simulations to study the structure and mechanical unfolding pathway of FN-III1. This study finds that FN-III1 consists of a beta-sandwich structure that unfolds to a mechanically stable intermediate which is about four times the length of the native folded state. Considering previous experimental findings, our studies provide a structural model by which mechanical stretching of FN-III1 may induce fibrillogenesis through this partially unfolded intermediate. |
|
Molecular dynamics investigation of primary photoinduced events in the activation of rhodopsin. Jan Saam, Emad Tajkhorshid, Shigehiko Hayashi, and Klaus Schulten. Biophysical Journal, 83:3097-3112, 2002 (Citations: 167) Retinal cis-trans isomerization and early relaxation steps have been studied in a 10ns molecular dynamics simulation of a fully hydrated model of membrane-embedded rhodopsin. The isomerization, induced by transiently switching the potential energy function governing the C |
 Molecular dynamics simulations of proteins in lipid bilayers.
James Gumbart, Yi Wang, Alekseij Aksimentiev, Emad Tajkhorshid, and Klaus Schulten.
Current Opinion in Structural Biology, 15:423-431, 2005.
Molecular dynamics simulations of proteins in lipid bilayers.
James Gumbart, Yi Wang, Alekseij Aksimentiev, Emad Tajkhorshid, and Klaus Schulten.
Current Opinion in Structural Biology, 15:423-431, 2005. (Citations: 166) With recent advances in X-ray crystallography of membrane proteins promising many new high-resolution structures, MD simulations become increasingly valuable for understanding membrane protein function, as they can unleash dynamic behavior concealed in the static structures. Dramatic increase in computational power in synergy with more efficient computational methodologies allows one today to carry out molecular dynamics simulations of any structurally known membrane protein in its native environment, covering the time scale of up to 0.1 |
 Neural network control of a pneumatic robot arm.
Ted Hesselroth, Kakali Sarkar, P. Patrick van der Smagt, and Klaus Schulten.
IEEE Transactions on Systems, Man, and Cybernetics, 24:28-37, 1994.
Neural network control of a pneumatic robot arm.
Ted Hesselroth, Kakali Sarkar, P. Patrick van der Smagt, and Klaus Schulten.
IEEE Transactions on Systems, Man, and Cybernetics, 24:28-37, 1994. (Citations: 161) A neural map algorithm has been employed to control a five-joint pneumatic robot arm and gripper through feedback from two video cameras. The pneumatically driven robot arm (SoftArm) employed in this investigation shares essential mechanical characteristics with skeletal muscle systems. To control the position of the arm, 200 neurons formed a network representing the three-dimensional workspace embedded in a four-dimensional system of coordinates from the two cameras, and learned a three-dimensional set of pressures corresponding to the end effector positions, as well as a set of 3 |
 Structural determinants of MscL gating studied by molecular dynamics simulations. Justin Gullingsrud, Dorina Kosztin, and Klaus Schulten. Biophysical Journal, 80:2074-2081, 2001 Structural determinants of MscL gating studied by molecular dynamics simulations. Justin Gullingsrud, Dorina Kosztin, and Klaus Schulten. Biophysical Journal, 80:2074-2081, 2001 (Citations: 163) The mechanosensitive channel of large conductance (MscL) in prokaryotes plays a crucial role in exocytosis as well as in the response to osmotic downshock. The channel can be gated by tension in the membrane bilayer. The determination of functionally important residues in MscL, patch-clamp studies of pressure-conductance relationships, and the recently elucidated crystal structure of MscL from Mycobaterium tuberculosis, have guided the search for the mechanism of MscL gating. Here, we present a molecular dynamics study of the MscL protein embedded in a fully hydrated POPC bilayer. Simulations totalling 3 ns in length were carried out under conditions of constant temperature and pressure using periodic boundary conditions and full electrostatics. The protein remained in the closed state corresponding to the crystal structure, as evidenced by its impermeability to water.. Analysis of equilibrium fluctuations showed that the protein was most immobile in the narrowest part of the channel. The gating process was investigated through simulations of the bare protein under conditions of constant surface tension. Under a range of conditions the transmembrane helices flattened as the pore widened. Implications for the gating mechanism in light of these and experimental results are discussed. |
Back to the main page


